![Esperoct® [antihemophilic factor (recombinant), glycopegylated-exei] logo](/content/dam/novonordisk/novomedlink/logos/bleeding-disorders/esperoct.svg)
Indicated for use in adults and children with hemophilia A for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes. Esperoct® is not indicated for the treatment of von Willebrand disease.
Protection across all age groups with individualized prophylaxis frequency1‑3
Get to know Esperoct® — a flexibility factor with the ability to individualize dosing frequency for your patients based on bleeding episodes.4
In patients aged 12 to 70 years
Long-term trial results confirm effective prophylaxis in patients (aged 12 to 70 years) for up to 6.6 years.1
A lower overall median ABR in adult and adolescent patients compared with the main phase was achieved1
Overall bleeds per year
N=177
Actor portrayal

Based on a post hoc analysis, the majority of adults/adolescents who completed the entire trial experienced no annual bleeds after year 1.1,b
bMedian ABR shown is from the main and extension phases of the pivotal clinical trial of PTPs aged ≥12 years with severe hemophilia A who received Esperoct® 50 IU/kg every 4 days. Patients who completed the entire pathfinder2 trial (n=110) who received Esperoct® 50 IU/kg every 4 days for up to 6.6 years. Patients evaluated at year 2 (n=103), year 3 (n=66), year 4 (n=62), year 5 (n=62), year 6 (n=59).1
Pivotal trial main phase data
Esperoct® provided effective prophylaxis with a low median ABR in all patients (aged 12 to 70 years).4
overall bleeds per year
(n=175)
spontaneous bleeds
traumatic bleeds
joint bleeds
ABR=annualized bleed rate.
Extension trial data: Individualized prophylaxis frequency based on bleeding episodes
Long-term use of Esperoct® in the pathfinder8 clinical trial resulted in protection from bleeds with a lower ABR.3,c,d
Post hoc subgroup analysis: results of twice-weekly dosing (pathfinder8)c
0 bleeds in second year of treatment3,c,d
of patients (n=61)
Mean ABR reduction3,c,e
in patients with an ABR >1 in pathfinder2 (n=30)
cA subgroup analysis was performed in 71 patients who switched from Esperoct® 50 IU/kg every 4 days in pathfinder2 to 50 IU/kg twice weekly in pathfinder8.3
dIn the subgroup analysis of 71 patients, 61 remained on 50 IU/kg twice-weekly dosing in the second year of pathfinder8.3
eIn the subgroup analysis, 30 of the 71 patients had a mean ABR >1 coming from pathfinder2. 90% of these patients demonstrated an improvement in ABR after transitioning to twice-weekly prophylaxis in pathfinder8. Three of these patients demonstrated an increase in ABR after transitioning to the new regimen, with a mean (SD) change in ABR of 2.67 (2.4).3
In previously treated patients aged 0 to <12 years
Long-term trial results confirm effective prophylaxis in children (aged 0 to <12 years) for up to 5.4 years.2
A lower overall median ABR in pediatric patients compared with the main phase was achieved2
100% resolution of target joints.2,f
Overall bleeds per year
N = 68
Actor portrayal

Based on a post hoc analysis of patients who completed the entire trial, the proportion of patients who experienced no annual bleeding episodes more than doubled from year 1 to year 5.2,g
fA target joint was defined as a single joint with ≥3 bleeding episodes in 6 consecutive months. All baseline target joints reached the per-protocol definition of target joint resolution in slightly over 2 years of treatment with Esperoct®. Per protocol, a target joint was no longer considered a target joint if there were no bleeding episodes for 12 consecutive months. Twelve patients with 16 documented target joints at the baseline participated in main and extension phase of pathfinder5 clinical trial.2
gPatients who completed the entire pathfinder5 trial who took Esperoct® 60 IU/kg (50 IU/kg – 75 IU/kg) twice weekly for a median of 4.9 years (n=63). Approximately 32% of the patients that participated in both the main and extension phases experienced no bleeding episodes during year 1, ~50% during year 2, <50% during year 3, 56% during year 4, and ~70% during year 5 had no annual bleeding episodes.2
Pivotal trial main phase data
Esperoct® achieved a low median ABR in children (aged 0 to <12 years).4
all bleeds
spontaneous bleeds
traumatic bleeds
joint bleeds
ABR=annualized bleed rate.
The pathfinder clinical trial program evaluated the efficacy and safety of Esperoct® in over 270 patients with hemophilia A worldwide.4
Esperoct® was shown to provide effective perioperative bleed control.
Trial prescription program

Qualifying patients can receive a trial prescription for Esperoct®.
To learn more about our trial prescription program, please call 1-844-668-6732 to speak with a NovoCare® Specialist.
Study designs
Giangrande et al (2017 and 2020)
pathfinder2 phase 3 clinical trial: main phase and extension
Patients: Previously treated males aged 12–66 years with severe congenital hemophilia A (FVIII activity <1%) and at least 150 exposure days to any FVIII products.5
Study design: Multinational, open-label, non-randomized trial evaluating safety, pharmacokinetics, and clinical efficacy of Esperoct® when used for prophylaxis and treatment of bleeds. During the main phase, 175 patients received routine prophylaxis (50 IU/kg every 4 days), and 12 adults elected to be treated on-demand. A subset of patients (n=150) continued on in extension phase part 1; 139 continued into the non-randomized extension part 2, with 113 completing the trial.1,5
Co-primary endpoints: Incidence of FVIII inhibitors ≥0.6 BU (Bethesda units) and ABR (annualized bleeding rate) for patients on a prophylaxis regimen.5
Lentz et al (2022)3
pathfinder8 phase 3 extension trial
Patients: Previously treated males (n=160) with severe hemophilia A (FVIII activity <1%) participating in pathfinder2 (n=102) and pathfinder5 (n=58) trials.
Study design: Multicenter, multinational, open-label, nonrandomized, interventional trial. Patients received Esperoct® prophylaxis dosed every 7 days (Q7D), twice weekly (BIW), or three times weekly (TIW), with dosing interval based on previous regimen and bleeding tendency. All patients on the Q7D regimen were aged ≥12 years, and the dose was 75 IU/kg. Dosing for BIW was 50 IU/kg (patients aged ≥12 years) or 60 IU/kg (patients aged <12 years). TIW dosing was 50 IU/kg (all ages).
Primary endpoint: Number of reported adverse events.
Secondary endpoints: Efficacy endpoints included number of treated bleeds on prophylaxis, pre-dose FVIII activity levels on prophylaxis, and treatment satisfaction. A secondary safety endpoint was the incidence of FVIII inhibitors.
Meunier et al (2017)6
pathfinder5 phase 3 clinical trial
Patients: Previously treated males (n=68) aged <12 years with severe hemophilia A (FVIII activity level of <1%) and a body weight ≥10 kg. Patients aged 0–5 years had at least 50 exposure days to other FVIII products. Patients aged 6–11 years had at least 150 exposure days.
Study design: Multinational, open-label, non-randomized, non-controlled trial evaluating the safety, clinical efficacy, and pharmacokinetics of prophylactic and on-demand use of Esperoct®. For bleeding episodes, treatment dose was 20–75 IU/kg, depending on bleed severity and location. For prophylaxis, patients administered 60 IU/kg (50–75 IU/kg) twice weekly for 26 weeks.
Prior to the start of routine prophylaxis, single-dose PK comparison was performed in 27 children between previous SHL products and Esperoct® at the same administered dose. Half-life comparison is based on estimated terminal half-life ratio using a population-based method.
Primary endpoint: Incidence of inhibitory antibodies against FVIII, indicated by a result of ≥0.6 BU, as measured with the Nijmegen-modified Bethesda assay with heat inactivation of residual FVIII activity.
Secondary endpoint: Frequency of adverse events, including inhibitor formation against FVIII, allergic reactions, thromboembolic events, and medication errors.
Šaulytė Trakymiene et al (2020)2
pathfinder5 phase 3 trial: extension phase
Patients: Previously treated males aged <12 years with severe hemophilia A who had participated in the pathfinder5 main phase (n=68).
Study design: Patients continued prophylactic use of Esperoct® at 60 IU/kg (50–75 IU/kg) twice or three times a week. ABR was estimated using a Poisson regression model with log (prophylaxis duration) as offset and estimating over-dispersion by Pearson's scale.
Primary endpoint: Incidence of inhibitory antibodies against Esperoct®.
Secondary endpoints: Frequency of adverse events, including inhibitor formation, allergic reactions, thromboembolic events, and medication errors.
Count on the clinical trial experience of Esperoct®.
PEGylation technology
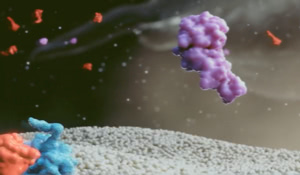
Esperoct® extends half-life through PEGylation.4,5,7
Important Safety Information for Esperoct®
Contraindications
- Do not use in patients who have known hypersensitivity to Esperoct® or its components, including hamster proteins
Warnings and Precautions
- Hypersensitivity reactions, including anaphylaxis, may occur. Should hypersensitivity reactions occur, discontinue Esperoct® and administer appropriate treatment
- Development of neutralizing antibodies (inhibitors) has occurred. Perform an assay that measures Factor VIII inhibitor concentration if bleeding is not controlled with the recommended dose of Esperoct® or if the expected plasma Factor VIII activity levels are not attained
- Temporary decrease in Factor VIII incremental recovery (IR) has been observed after Esperoct® infusion, within the first 5 exposure days, in previously untreated patients (PUPs) <6 years of age. During the decreased IR period, these subjects may have an increased bleeding tendency. If bleeding is not controlled with the recommended dose of Esperoct® and/or the expected Factor VIII activity levels are not attained and Factor VIII inhibitors are not detected, consider adjusting the dose, dosing frequency, or discontinuing Esperoct®
Adverse Reactions
- The most frequently reported adverse reactions in clinical trials (≥1%) were rash, redness, itching (pruritus), and injection site reactions. Additional frequently reported adverse reactions (≥1%) in PUPs included Factor VIII inhibition and hypersensitivity.
Please click here for Esperoct® Prescribing Information.
Indications and Usage
Esperoct® [antihemophilic factor (recombinant), glycopegylated-exei] is indicated for use in adults and children with hemophilia A for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes
- Esperoct® is not indicated for the treatment of von Willebrand disease
Important Safety Information for Esperoct®
Contraindications
- Do not use in patients who have known hypersensitivity to Esperoct® or its components, including hamster proteins
Warnings and Precautions
- Hypersensitivity reactions, including anaphylaxis, may occur. Should hypersensitivity reactions occur, discontinue Esperoct® and administer appropriate treatment
- Development of neutralizing antibodies (inhibitors) has occurred. Perform an assay that measures Factor VIII inhibitor concentration if bleeding is not controlled with the recommended dose of Esperoct® or if the expected plasma Factor VIII activity levels are not attained
- Temporary decrease in Factor VIII incremental recovery (IR) has been observed after Esperoct® infusion, within the first 5 exposure days, in previously untreated patients (PUPs) <6 years of age. During the decreased IR period, these subjects may have an increased bleeding tendency. If bleeding is not controlled with the recommended dose of Esperoct® and/or the expected Factor VIII activity levels are not attained and Factor VIII inhibitors are not detected, consider adjusting the dose, dosing frequency, or discontinuing Esperoct®
Adverse Reactions
- The most frequently reported adverse reactions in clinical trials (≥1%) were rash, redness, itching (pruritus), and injection site reactions. Additional frequently reported adverse reactions (≥1%) in PUPs included Factor VIII inhibition and hypersensitivity.
Please click here for Esperoct® Prescribing Information.
Indications and Usage
Esperoct® [antihemophilic factor (recombinant), glycopegylated-exei] is indicated for use in adults and children with hemophilia A for on-demand treatment and control of bleeding episodes, perioperative management of bleeding, and routine prophylaxis to reduce the frequency of bleeding episodes
- Esperoct® is not indicated for the treatment of von Willebrand disease
References:
- Giangrande P, Karim F, Nemes L, et al. Long-term safety and efficacy of N8-GP in previously treated adults and adolescents with hemophilia A: final results from pathfinder2. J Thromb Haemost. 2020;18(1):5-14.
- Šaulytė Trakymiene S, Economou M, Kenet G, Landorph A, Shen C, Kearney S. Long-term safety and efficacy of N8-GP in previously treated pediatric patients with hemophilia A: final results from pathfinder5. J Thromb Haemost. 2020;18(suppl 1):15-25.
- Lentz SR, Kavalki K, Klamroth R, et al. Turoctocog alpha pegol (NB-GP) in severe hemophilia A: long-term safety and efficacy in previously treated patients of all ages in the pathfinder8 study. Res Pract Thromb Haemost. 2022;6(2):e12674.
- Esperoct® [package insert]. Plainsboro, NJ: Novo Nordisk Inc.
- Giangrande P, Andreeva T, Chowdary P, et al. Clinical evaluation of glycoPEGylated recombinant FVIII: efficacy and safety in severe haemophilia A. Thromb Haemost. 2017;117(2):252-261.
- Meunier S, Alamelu J, Ehrenforth S, et al. Safety and efficacy of a glycoPEGylated rFVIII (turoctocog alpha pegol, N8-GP) in paediatric patients with severe haemophilia A. Thromb Haemost. 2017;117:1705-1713.
- Tiede A, Brand B, Fischer R, et al. Enhancing the pharmacokinetic properties of recombinant factor VIII: first-in-human trial of glycoPEGylated recombinant factor VIII in patients with hemophilia A. J Thromb Haemost. 2013;11(4):670-678.